
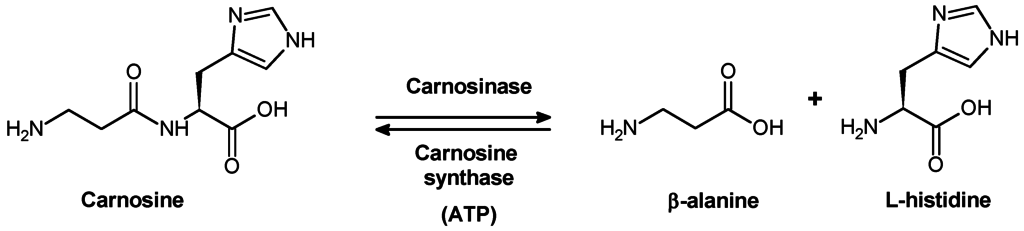
It is necessary to clarify the metabolic pathways of carnosine conversion in organism to understand the biological significance of this compound. The absence of carnosine in urine under moderate exercise training (Hunter, 1925) suggests, that normally unmodified carnosine is not excreted from human body, but it is subject to metabolic degradation before excretion. It was in 1915 that Dietrich showed that neither pepsin nor trypsin digests carnosine molecule. Therefore, it was important to reveal which enzyme is responsible for carnosine hydrolysis.
Skeletal muscles are the main depot of carnosine and anserine. Therefore, it was quite natural to expect that metabolic conversion of these dipeptides is associated with vital activities of muscle cells. Meshkova and Zolotarevskaya concluded that carnosine neither undergoes autolytic transformation in muscular tissue nor is hydrolysed by muscular enzymes (Meshkova, Zolotarevskaya, 1937). It was shown in the laboratory of S.E. Severin that kidney, liver, spleen, and erythrocytes contain an enzyme capable of catalysing hydrolysis of carnosine (Гаркави, 1938, 1940; Северин, Георгиевска, 1938). On the basis of structural characteristics of this enzyme, Garkavi (Гаркави, 1938) suggested that it is carboxypolypeptidase. An enzyme capable of hydrolyzing carnosine was isolated in 1949 from pig kidney (Hanson and Smith, 1949). Manganese ions and cyanides or sulphides caused activation and inhibition of the activity of the enzyme, respectively. In addition to carnosine, this enzyme catalysed hydrolysis of some other dipeptides but at much slower rate.
When pig kidney carnosinase bad been purified for the first time, it was found that in the absence of metal ions this enzyme is highly substrate-specific, because it hydrolyses carnosine and anserine rather than homocarnosine. In the presence of Co2+, carnosinase loses substrate specificity and catalyses hydrolysis of homocarnosine and many other dipeptides (Lenney, 1990). The molecular weight of the enzyme is 57 kDa; pl 5.5; and Michaelis constant value (Km) for carnosine is lower 0.4 mM. Carnosinase is highly selective to carnosine. Because even small modifications of dipeptide structure cause a significant effect on the activity of the enzyme, anserine is considered as “bad” substrate, whereas homocarnosine is not hydrolysed at all.
Carnosinase is heterogeneously distributed over the whole body and over different structures of animal brain. Human body tissues also contain two types of dipeptidases capable of hydrolysing carnosine. These enzymes substantially differ from one another in activation by metal ions, molecular weight, substrate specificity, etc. One of these enzymes, “tissue” carnosinase is not able to hydrolase neither homocarnosine nor anserine. The second enzyme is able to hydrolyse carnosine, anserine, and, to a lesser extent, homocarnosine. This enzyme is abundant in blood serum, where it plays an important role in hydrolysis of dietary carnosine ( “serum” carnosinase)
Reference:
Schoen, P., Everts, H., de Boer, T., & van Oeveren, W. (2003). Serum carnosinase activity in plasma and serum: validation of a method and values in cardiopulmonary bypass surgery. Clinical chemistry, 49(11), 1930-1932.