
Il est nécessaire de clarifier les voies métaboliques de la conversion de carnosine dans l’organisme pour comprendre la signification biologique de ce composé. L’absence de carnosine dans l’urine sous un entraînement physique modéré (Hunter, 1925) suggère que la carnosine normalement non modifiée n’est pas excrétée du corps humain, mais qu’elle est soumise à une dégradation métabolique avant l’excrétion. C’est en 1915 que Ditrich a montré que ni la pepsine ni la trypsine ne digèrent la molécule de carnosine. Par conséquent, il était important de révéler quelle enzyme est responsable de l’hydrolyse de la carnosine.
Les muscles squelettiques sont le principal dépôt de carnosine et d’ansérine. Par conséquent, il était tout à fait naturel de s’attendre à ce que la conversion métabolique de ces dipeptides soit associée à des activités vitales des cellules musculaires. Meshkova et Zolotarevskaya ont conclu que la carnosine ne subit pas de transformation auto lytique dans le tissu musculaire et qu’elle n’est pas hydrolysée par les enzymes musculaires (Meshkova, Zolotarevskaya, 1937). Il a été montré dans le laboratoire de S.E. Severin que le rein, le foie, la rate et les érythrocytes contiennent une enzyme capable de catalyser l’hydrolyse de la carnosine (Гаркави, 1938, 1940; Северин, Георгиевска, 1938). Sur la base des caractéristiques structurales de cette enzyme, Garkavi (Гаркави, 1938) a suggéré que c’est la carboxypolypeptidase. Une enzyme capable d’hydrolyser la carnosine a été isolée en 1949 à partir de rein de porc (Hanson et Smith, 1949). Les ions manganèse et les cyanures ou sulfures provoquent respectivement l’activation et l’inhibition de l’activité de l’enzyme. En plus de la carnosine, cette enzyme a catalysé l’hydrolyse de certains autres dipeptides mais à un rythme beaucoup plus lent.
Lorsque la carnosinase rénale du porc a été purifiée pour la première fois, on a constaté que, en l’absence d’ions métalliques, cette enzyme est très spécifique du substrat, car elle hydrolyse la carnosine et l’ansérine plutôt que l’homocarnosine. En présence de Co2 +, la carnosinase perd sa spécificité de substrat et catalyse l’hydrolyse de l’homocarnosine et de nombreux autres dipeptides (Lenney, 1990). Le poids moléculaire de l’enzyme est de 57 kDa; pl 5,5; et la valeur constante de Michaelis (Km) pour la carnosine est inférieure à 0,4 mM. La carnosinase est hautement sélective pour la carnosine. Parce que même de petites modifications de la structure di peptidique provoquent un effet significatif sur l’activité de l’enzyme, l’ansérine est considérée comme un “mauvais” substrat, alors que l’homocarnosine n’est pas du tout hydrolysée.
La carnosinase est distribuée de manière hétérogène sur tout le corps et sur différentes structures du cerveau animal. Les tissus du corps humain contiennent également deux types de di peptidases capables d’hydrolyser la carnosine. Ces enzymes diffèrent sensiblement l’une de l’autre par l’activation par les ions métalliques, le poids moléculaire, la spécificité du substrat, etc. L’une de ces enzymes, la carnosinase “tissulaire”, n’est pas capable d’hydrolyser ni l’homocarnosine ni l’ansérine. La seconde enzyme est capable d’hydrolyser la carnosine, l’ansérine et, dans une moindre mesure, l’homocarnosine. Cette enzyme est abondante dans le sérum sanguin, où elle joue un rôle important dans l’hydrolyse de la carnosine alimentaire (“sérum” carnosinase)
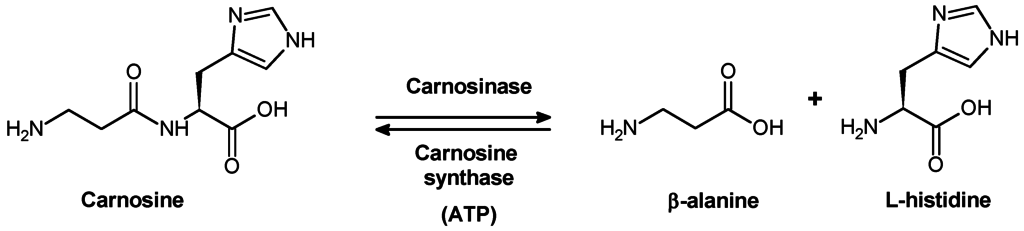
La carnosinémie, également appelée carence en carnosinase, est un trouble métabolique autosomalrécessif rare causé par une carence en carnosinase, une di peptidase (un type d’enzyme qui divise les dipeptides en deux constituants des acides aminés). Le gène de la carnosinase est situé sur le chromosome 18, autosome. . Le gène de la carnosine dipeptidase-1 contrôle les carnosinases tissulaires et sériques. Les mutations de ce gène sont responsables d’une carence en carnosinase, entraînant une carnosinémie. La carence en carnosinase sérique, avec la carnosinurie (“carnosine dans l’urine”), est l’indicateur métabolique habituel de la carence en carnosinase systémique. Cette forme de la di peptidase n’est pas trouvée dans le sang humain jusqu’à la fin de la petite enfance, atteignant lentement l’âge adulte à l’âge de 15 ans. Une variété de symptômes neurologiques ont été associés à la carnosinémie. Ils comprennent: hypotonie, retard de développement, retard mental, dégénérescence des axones, neuropathie sensorielle, tremblements, démyélinisation, anomalies de la substance grise et crises myocloniques.